Hipertensão pulmonar
A hipertensão pulmonar ou PH é uma condição em que há hipertensão arterial nos pulmões. Esta condição torna difícil a respiração. Algumas pessoas com esta condição precisam de oxigênio extra. Esta condição também pode deixar uma pessoa tonta e f…
A hipertensão pulmonar ou PH é uma condição em que há hipertensão arterial nos pulmões. Esta condição torna difícil a respiração. Algumas pessoas com esta condição precisam de oxigênio extra. Esta condição também pode deixar uma pessoa tonta e ficar facilmente cansada. Algumas pessoas com esta condição desmaiam facilmente. Os sintomas se agravam quando se exercita ou trabalha duro. A hipertensão pulmonar é uma condição grave, e pode ser fatal. A condição faz com que seja mais difícil para o coração bombear sangue. Como o coração tem que trabalhar mais, ele também pode adoecer. Algumas pessoas que estão muito doentes podem precisar de um transplante pulmonar ou de um transplante de coração-pulmão para viver. Hipertensão pulmonar nome completo é hipertensão arterial pulmonar, embora a maioria das pessoas a chame de pah, ph ou pha.
Galeria de imagens
9 Imagens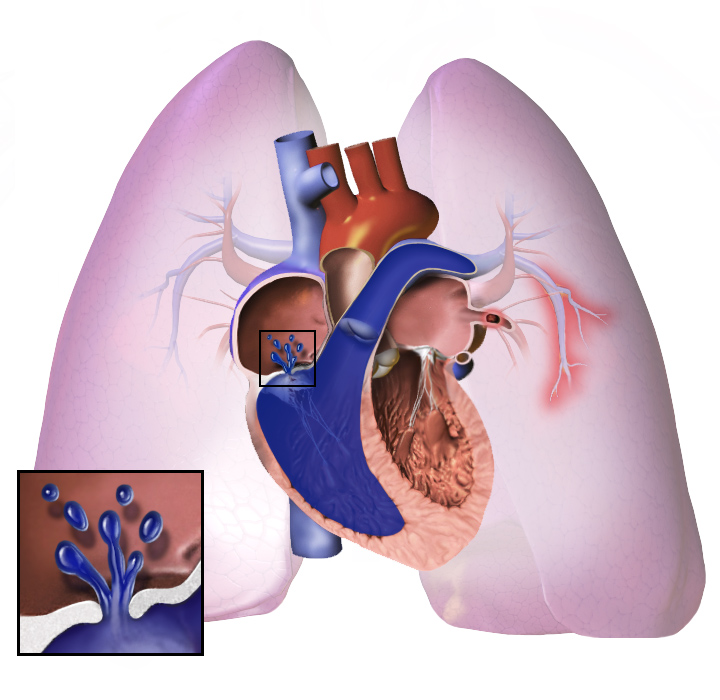






Sinais e sintomas
As pessoas com hipertensão pulmonar têm dificuldade de respirar. Elas também se cansam facilmente. Algumas delas também desmaiam com facilidade. Elas podem ter dores no peito. Alguns pacientes têm inchaço dos pés e dos tornozelos. Estes sintomas se agravam durante o exercício ou o trabalho duro.
Como muitas doenças podem dificultar a respiração, um médico deve aprender sobre os antecedentes do paciente. Isto ajuda o médico a tratar o paciente, mesmo se o paciente tiver outra doença. O médico também faz vários testes. A hipertensão pulmonar faz o coração parecer diferente. Um teste é medir a pressão sanguínea dentro da artéria pulmonar, o vaso sanguíneo que vai do coração para os pulmões.
A fim de estabelecer a causa, o médico geralmente conduzirá um histórico médico completo. Um histórico familiar detalhado é levado em consideração para determinar se a doença pode ser familiar. Um histórico de exposição à cocaína, metanfetamina, álcool levando à cirrose e fumo levando ao enfisema são considerados significativos. O exame físico é realizado para procurar sinais típicos de hipertensão pulmonar, incluindo um alto P2 (som de fechamento da válvula pulmonar), (para)cavalete esternal, distensão venosa jugular, edema de pedal, ascite, refluxo hepatojugular, baqueteamento, etc.
O que vai mal com o corpo
Na hipertensão pulmonar, os vasos sanguíneos dos pulmões tornam-se muito estreitos. A pressão sanguínea nos pulmões torna-se alta. O coração trabalha muito para bombear o sangue através dos vasos sanguíneos estreitos. Mais tarde, os vasos sanguíneos nos pulmões tornam-se duros e espessos. O coração deve trabalhar com mais afinco.
O coração pode trabalhar tanto que fica doente. Isto é chamado de insuficiência cardíaca. O coração doente não pode bombear bem o sangue. Menos sangue vai para os pulmões, então o sangue recebe menos oxigênio. Isto faz com que seja difícil respirar. Isto fica pior quando se exercita ou trabalha duro.
Causas
A causa mais comum da hipertensão pulmonar é a insuficiência cardíaca esquerda. Isto causa hipertensão venosa pulmonar. Isto leva a edema pulmonar, ou acúmulo de líquido nos pulmões.
Muitas doenças podem causar hipertensão arterial pulmonar (HAP).
- Doenças pulmonares que fazem com que o sangue tenha menos oxigênio, como por exemplo:
· doença pulmonar obstrutiva crônica ou DPOC
· doença pulmonar intersticial
· Síndrome de Pickwickian
- problemas do sistema imunológico, tais como:
· AIDS
· scleroderma
· outras doenças auto-imunes
- problemas de fígado
· cirrose
· hipertensão portal
- outras causas
· apnéia do sono
· tomar comprimidos para perder peso, tais como Fen-Phen, Aminorex, fenfluramina (Pondimin), e fentermina
· doença falciforme,
· cardiopatia congênita
· doenças da tireóide,
· tomando drogas como cocaína
· possivelmente herpesvírus humano 8
Quando uma pessoa tem hipertensão pulmonar sem qualquer outra causa, isto é chamado de hipertensão arterial pulmonar idiopática ou IPAH.
Quando existe um histórico familiar, a doença é denominada hipertensão arterial pulmonar familiar, (FPAH). O IPAH e a FPAH são agora considerados distúrbios genéticos ligados a mutações no gene BMPR2, que codifica um receptor para proteínas morfogenéticas ósseas, assim como o gene 5-HT(2B), que codifica um receptor de serotonina.
Na medicina, a hipertensão pulmonar (HP) é um aumento da pressão arterial na artéria pulmonar ou vasculatura pulmonar, levando à falta de ar, tonturas, desmaios e outros sintomas, todos eles exacerbados pelo esforço. Dependendo da causa, a hipertensão pulmonar pode ser uma doença grave com uma tolerância ao exercício acentuadamente reduzida e insuficiência cardíaca do lado direito. Ela foi identificada pela primeira vez pelo Dr. Ernst von Romberg em 1891. Pode ser de cinco tipos diferentes, arterial, venosa, hipóxica, tromboembólica, ou mista.
Embora os termos hipertensão pulmonar primária (significado de causa desconhecida) e hipertensão pulmonar secundária (significado devido a outra condição médica) ainda persistam em materiais divulgados aos pacientes e ao público em geral, estes termos têm sido largamente abandonados na literatura médica. Esta mudança ocorreu porque a classificação dicotômica mais antiga não refletia a fisiopatologia ou o resultado. Ela levou a decisões terapêuticas errôneas, ou seja, tratar apenas a hipertensão pulmonar "primária". Isto, por sua vez, levou ao niilismo terapêutico para muitos pacientes rotulados como hipertensão pulmonar "secundária", e poderia ter contribuído para suas mortes. O termo "hipertensão pulmonar primária" foi agora substituído por "hipertensão arterial pulmonar idiopática". Os termos "hipertensão pulmonar primária" e "secundária" não devem mais ser utilizados. Mais detalhes estão na seção de Classificação abaixo.
Causas
A causa mais comum da hipertensão pulmonar é a insuficiênciacardíaca esquerda que leva à hipertensão venosa pulmonar. Isto pode ser devido ao mau funcionamento sistólico ou diastólico do ventrículo esquerdo ou devido a disfunção valvar, como regurgitação mitral ou estenose mitral. Geralmente se manifesta como um edema pulmonar.
As causas comuns de hipertensão arterial pulmonar (HAP) incluem HIV, escleroderma e outros distúrbios auto-imunes, cirrose e hipertensão portal, doença falciforme, doença cardíaca congênita, doenças da tireóide e outras. O uso de comprimidos de emagrecimento como o Fen-Phen, Aminorex, fenfluramina (Pondimin) e fentermina levou ao desenvolvimento da HAP no passado.
Patogênese
Qualquer que seja a causa inicial, a hipertensão pulmonar envolve o aperto dos vasos sanguíneos conectados aos pulmões e dentro deles. Isto torna mais difícil para o coração bombear o sangue através dos pulmões, assim como é mais difícil fazer a água fluir através de um tubo estreito em oposição a um tubo largo. Com o tempo, os vasos sanguíneos afetados tornam-se mais rígidos e mais espessos, aumentando ainda mais a pressão sanguínea dentro dos pulmões e prejudicando o fluxo sanguíneo. Além disso, o aumento da carga de trabalho do coração causa espessamento e alargamento do ventrículo direito, tornando o coração menos capaz de bombear o sangue através dos pulmões, causando a insuficiência cardíaca direita. À medida que o sangue que flui pelos pulmões diminui, o lado esquerdo do coração recebe menos sangue. Este sangue também pode transportar menos oxigênio do que o normal. Portanto, torna-se cada vez mais difícil para o lado esquerdo do coração bombear para fornecer oxigênio suficiente para o resto do corpo, especialmente durante a atividade física.
Diagnóstico
Como a hipertensão pulmonar pode ser de 5 tipos principais, uma série de testes deve ser realizada para distinguir a hipertensão arterial pulmonar de variedades venosas, hipóxicas, tomboembólicas ou miscelâneas.
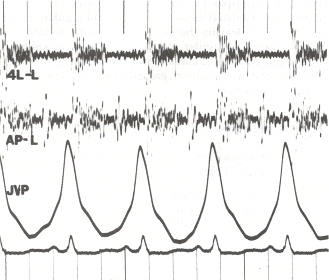
Um exame físico é realizado para procurar sinais típicos de hipertensão pulmonar. Estes incluem sons cardíacos alterados, tais como um S2 amplamente dividido ou segundo som cardíaco, um P2 alto ou som de fechamento da válvula pulmonar (parte do segundo som cardíaco), (para) heave esternal, possível som S3 ou terceiro som cardíaco, e regurgitação pulmonar. Outros sinais incluem distensão venosa jugular (aumento das veias jugulares), edema periférico (inchaço dos tornozelos e pés), ascite (inchaço abdominal devido ao acúmulo de líquido), refluxo hepatojugular, e baqueamento.
São necessários procedimentos adicionais para confirmar a presença de hipertensão pulmonar e excluir outros diagnósticos possíveis. Estes geralmente incluem testes de função pulmonar, exames de sangue, eletrocardiografia (ECG), medidas de gases no sangue arterial, radiografias do tórax (seguidas de tomografia computadorizada de alta resolução se houver suspeita de doença pulmonar intersticial), e ventilação-perfusão ou varredura V/Q para excluir hipertensão pulmonar tromboembólica crônica. A biópsia do pulmão geralmente não é indicada a menos que se pense que a hipertensão pulmonar seja devida a uma doença pulmonar intersticial subjacente. Mas as biópsias pulmonares estão repletas de riscos de sangramento devido à alta pressão sanguínea intrapulmonar. A melhora clínica é freqüentemente medida por um "teste de caminhada de seis minutos", ou seja, a distância que um paciente pode caminhar em seis minutos. A estabilidade e a melhora nesta medida estão correlacionadas com uma melhor sobrevivência.
Embora a pressão arterial pulmonar possa ser estimada com base na ecocardiografia, a amostragem de pressão com um cateter de Swan-Ganz fornece a medida mais definitiva. PAOP e PVR não podem ser medidos diretamente com o ecocardiograma. Portanto, o diagnóstico de HAP requer um cateterismo cardíaco. Um cateter de Swan-Ganz também pode medir o débito cardíaco, o que é muito mais importante para medir a gravidade da doença do que a pressão arterial pulmonar.
A pressão arterial pulmonar normal em uma pessoa que vive ao nível do mar tem um valor médio de 12-16 mm Hg (1600-2100 Pa). A hipertensão pulmonar definida está presente quando as pressões médias em repouso excedem 25 mm Hg (3300 Pa). Se a pressão média da artéria pulmonar aumentar acima de 30 mm Hg (4000 Pa) com exercício, isso também é considerado hipertensão pulmonar.
O diagnóstico de HAP requer a presença de hipertensão pulmonar com duas outras condições. A pressão de oclusão da artéria pulmonar (PAOP ou PCWP) deve ser inferior a 15 mm Hg (2000 Pa) e a resistência vascular pulmonar (PVR) deve ser superior a 3 unidades de madeira (240 dyn-s-cm-5 ou 2,4 mN-s-cm-5).
Classificação
Classificação atual
Em 2003, o 3º Simpósio Mundial sobre Hipertensão Arterial Pulmonar foi convocado em Veneza para modificar a classificação com base no novo entendimento dos mecanismos da doença. O sistema revisado desenvolvido por este grupo fornece a estrutura atual para a compreensão da hipertensão pulmonar.
O sistema inclui várias melhorias em relação ao antigo sistema de Classificação de Evian de 1998. As descrições dos fatores de risco foram atualizadas, e a classificação de shunts sistêmico-pulmonares congênitos foi revista. Uma nova classificação de fatores genéticos na HP foi recomendada, mas não implementada porque os dados disponíveis foram considerados inadequados.
O sistema de Classificação Revisada de Veneza 2003 pode ser resumido como se segue:
- OMS Grupo I - Hipertensão arterial pulmonar (HAP)
- OMS Grupo II - Hipertensão pulmonar associada à cardiopatia esquerda
- Grupo III da OMS - Hipertensão pulmonar associada a doenças pulmonares e/ou hipoxemia
- Grupo IV da OMS - Hipertensão pulmonar devido a doença trombótica crônica e/ou embólica
- OMS Grupo V - Diversos
Terminologia anterior
Os termos hipertensão pulmonar primária e secundária (HPP e HPS) foram usados anteriormente para classificar a doença. Isto levou à suposição de que apenas a doença primária deveria ser tratada, e a variedade secundária deveria ser ignorada em favor do tratamento apenas da doença subjacente. De fato, todas as formas de hipertensão arterial pulmonar são tratáveis. Infelizmente, este sistema de classificação ainda persiste na mente de muitos médicos, e provavelmente leva a muitos pacientes com o tratamento negado. Esta abordagem niilista da hipertensão arterial pulmonar também pode contribuir para o sub-diagnóstico. Estima-se que existam cerca de 100.000 pacientes com HAP nos EUA, mas apenas 15-20.000 foram diagnosticados. Muitos outros foram erroneamente diagnosticados como DPOC, asma, ou insuficiência cardíaca congestiva.
O termo hipertensão pulmonar primária (HPP) foi agora substituído por hipertensão arterial pulmonar idiopática (HAP) em grande parte da literatura médica. No entanto, alguns médicos continuam a utilizar a classificação mais antiga de forma inadequada.
Epidemiologia
O IPAH é uma doença rara com uma incidência de cerca de 2-3 por milhão por ano e uma prevalência de cerca de 15 por milhão. As mulheres são quase três vezes mais propensas a se apresentar com o IPAH do que os homens.
Outras formas de HAP são muito mais comuns. Na esclerodermia, a incidência foi estimada em 6 a 60% de todos os pacientes, na artrite reumatóide até 21%, no lúpus eritematoso sistêmico 4 a 14%, na hipertensão portal entre 2 e 5%, no HIV cerca de 0,5%, e na doença falciforme que varia de 20 a 40%.
Comprimidos dietéticos como o Fen-Phen produziram uma incidência anual de 25-50 por milhão por ano.
Tratamento
O tratamento é determinado por se o PH é arterial, venoso, hipóxico, tromboembólico ou variado. Como hipertensão venosa pulmonar é sinônimo de insuficiência cardíaca congestiva, o tratamento é para otimizar a função ventricular esquerda pelo uso de diuréticos, beta bloqueadores, inibidores da ECA, etc., ou para reparar/substituir a válvula mitral ou aórtica.
Na HAP, mudanças no estilo de vida, digoxina, diuréticos, anticoagulantes orais e oxigenoterapia são consideradas terapia convencional, mas nunca foram comprovadas como benéficas de forma aleatória e prospectiva.
Os bloqueadores de canais de cálcio em altas doses são úteis em apenas 5% dos pacientes IPAH que são vasoreativos pelo cateter de Swan-Ganz. Infelizmente, os bloqueadores dos canais de cálcio têm sido amplamente mal utilizados, sendo prescritos a muitos pacientes com HAP não vasoreativos, levando a um excesso de morbidade e mortalidade.
Substâncias vasoativas
Três vias principais estão envolvidas na proliferação anormal e contração das células de músculo liso da artéria pulmonar em pacientes com hipertensão arterial pulmonar. Estas vias correspondem a alvos terapêuticos importantes nesta condição e desempenham um papel na determinação de quais das três classes de drogas - antagonistas dos receptores de endotelina, inibidores da fosfodiesterase tipo 5 e derivados da prostaciclina - serão utilizadas.
Prostacyclin (prostaglandina I2) é geralmente considerado o tratamento mais eficaz para a HAP. O Epoprostenol (prostacyclin sintético, comercializado como Flolan®) é dado através de infusão contínua que requer um cateter venoso central semi-permanente. Este sistema de administração pode causar sepse e trombose. Flolan® é instável e, portanto, tem que ser mantido em gelo durante a administração. Como tem uma meia-vida de 3 a 5 minutos, a infusão tem que ser contínua (24/7), e a interrupção pode ser fatal. Outros prostanóides, portanto, foram desenvolvidos. O treprostinil (Remodulin®) pode ser administrado por via intravenosa ou subcutânea, mas a forma subcutânea pode ser muito dolorosa. O Iloprost (Ilomedin®) também é usado na Europa por via intravenosa e tem uma meia-vida mais longa. Aloprost (comercializada como Ventavis®) é a única forma inalada de prostaciclina aprovada para uso nos EUA e Europa. Esta forma de administração tem a vantagem de depósito seletivo nos pulmões com menos efeitos colaterais sistêmicos.
O bosentan dual (ETA e ETB) antagonista do receptor de endotelina (comercializado como Tracleer®) foi aprovado em 2001. Dois antagonistas seletivos do receptor de endotelina (apenas ETA) estão em fase final de aprovação: sitaxsentan e ambrisentan. Sildenafil, um inibidor seletivo da fosfodiesterase específica de cGMP tipo 5 (PDE5), foi aprovado para o tratamento de PAH em 2005. Ele é comercializado para PAH como Revatio®. O Tadalafil (atualmente comercializado como Cialis® para disfunção erétil) é atualmente um ensaio de Fase III. O peptídeo intestinal vasoativo por inalação deve entrar em testes clínicos para HAP em 2007. PRX-08066 é um antagonista da serotonina atualmente sendo desenvolvido para hipertensão pulmonar hipóxica.
Cirúrgico
A septoestomia atrial é um procedimento cirúrgico que cria uma comunicação entre os átrios direito e esquerdo. Ela alivia a pressão no lado direito do coração, mas ao custo de níveis mais baixos de oxigênio no sangue (hipóxia). Ela é melhor realizada em centros experientes. O transplante pulmonar cura a hipertensão arterial pulmonar, mas deixa o paciente com as complicações do transplante, e uma sobrevida de cerca de 5 anos.
A tromboendarterectomia pulmonar pulmonar (TEP) é um procedimento cirúrgico utilizado para hipertensão pulmonar tromboembólica crônica. É a remoção cirúrgica de um trombo organizado (coágulo) junto com o revestimento da artéria pulmonar; é um procedimento grande e muito difícil que atualmente é realizado em alguns centros selecionados. A série de casos mostra um sucesso notável na maioria dos pacientes.
O tratamento para hipóxicos e variedades diversas de hipertensão pulmonar não foram estabelecidos. Entretanto, estudos de vários agentes estão atualmente matriculando pacientes. Muitos médicos tratarão estas doenças com os mesmos medicamentos que para a HAP, até que melhores opções se tornem disponíveis.
Prognóstico
O registro do NIH IPAH dos anos 80 mostrou uma sobrevida mediana não tratada de 2-3 anos a partir do momento do diagnóstico, sendo a causa de morte geralmente a falha ventricular direita (cor pulmonale). Embora este número seja amplamente citado, é provavelmente irrelevante hoje em dia. Os resultados mudaram drasticamente durante as duas últimas décadas. Isto pode ser devido a uma nova terapia medicamentosa, melhores cuidados gerais e diagnóstico mais precoce (tendência de lead time). Um estudo recente dos resultados dos pacientes que começaram o tratamento com bosentan (Tracleer®) mostrou que 86% dos pacientes estavam vivos aos 3 anos. Com vários agentes agora disponíveis, a terapia combinada é cada vez mais utilizada. O impacto destes agentes na sobrevivência não é conhecido, uma vez que muitos deles foram desenvolvidos apenas recentemente. Não seria irrazoável esperar que a mediana de sobrevivência se estendesse nos últimos 10 anos num futuro próximo.
Perguntas e respostas
P: O que é a hipertensão pulmonar ou PH?
R: A hipertensão pulmonar ou PH é uma doença em que existe uma pressão sanguínea elevada nos pulmões.
P: Quais são os sintomas da hipertensão pulmonar?
R: Os sintomas da hipertensão pulmonar incluem dificuldade em respirar, tonturas, fadiga e desmaios.
P: Porque é que algumas pessoas com hipertensão pulmonar precisam de mais oxigénio?
R: Algumas pessoas com hipertensão pulmonar precisam de oxigénio suplementar porque a doença lhes dificulta a respiração.
P: Quando é que os sintomas da hipertensão pulmonar se agravam?
R: Os sintomas da hipertensão pulmonar agravam-se quando faz exercício ou trabalha muito.
P: Porque é que a hipertensão pulmonar é uma doença grave?
R: A hipertensão pulmonar é uma doença grave porque dificulta o bombeamento de sangue pelo coração e pode ser fatal.
P: Qual é o nome completo da hipertensão pulmonar?
R: O nome completo da hipertensão pulmonar é hipertensão arterial pulmonar, embora a maioria das pessoas lhe chame pah, ph ou pha.
P: O que é que algumas pessoas muito doentes com hipertensão pulmonar precisam para viver?
R: Algumas pessoas muito doentes com hipertensão pulmonar podem precisar de um transplante de pulmão ou de um transplante coração-pulmão para viver.
Artigos relacionados
Autor
AlegsaOnline.com Hipertensão pulmonar Leandro Alegsa
URL: https://pt.alegsaonline.com/art/80022
Fontes
- ncbi.nlm.nih.gov : PMID 8692238
- ncbi.nlm.nih.gov : PMID 14985486
- ncbi.nlm.nih.gov : PMID 10555089
- ncbi.nlm.nih.gov : PMID 13679525
- ncbi.nlm.nih.gov : PMID 10903931
- ncbi.nlm.nih.gov : PMID 14659797
- ncbi.nlm.nih.gov : PMID 15194171